Calcium imaging#
Extracellular electrophysiology is just one of many methods that is used to measure activity from individual neurons. Another class of methods involve taking optical measurements of fluorescent compounds that change their properties in response to action potentials. One class of compounds changes fluorescence as the membrane voltage changes. These are called voltage indicators and provide access to the same signal that intracellular electrodes measure. However, the field is still developing voltage indicators that have good optical properties, and as a result these are not yet routinely used to measure neural activity in awake behaving animals. Another class uses the fact that action potentials trigger voltage-gated calcium channels (VGCCs) to open and increase the intracellular concentration of calcium. Unlike voltage indicators, there are many robust calcium indicators that provide good sensitivity and signal-to-noise ratio (SNR). These allow experimenters to observe action potentials using fluorescence microscopy.
Fluorescence: translating calcium into light#
Calcium-sensitive chemicals (fluorophores), which change their fluorescence properties upon
binding to Ca2+, are central to calcium imaging. Fluorophores emit light in a
characteristic wavelength while being excited by a light source. There are generally two
types used in calcium imaging; fluorescent dyes and proteins called {term}
genetically-encoded calcium indicators (GECIs). Dyes are chemicals which are
manually introduced into the cell in question, while GECIs are naturally produced by a
transgenic line. These indicators are introduced into neurons, resulting in the cells glowing
when under a fluorescence microscope; the glow changes as calcium concentrations in their
environments change. We can thus obtain a dynamic picture of neural activity across
populations of neurons by recording the fluorescence over time, providing insights into how
neural ensembles encode, process, and transmit information.
Advantages in the context of systems neuroscience#
Granular view: Calcium imaging enables a detailed view of large neural populations, capturing the dynamics of hundreds to thousands of neurons simultaneously. Better yet, some microscopes can resolve dendrites, individual synapses, or other cellular compartments.
Cell-type specificity: Thanks to advances in molecular biology, we can target GECIs to specific neuron types, granting us the power to dissect neural circuits with cell-type precision.
Longitudinal recordings: Because calcium imaging allows experimenters to see the shape
of individual neurons as well as their arrangement with respect to each other and other tissue features (like vasculature), experimenters can measure the activity of the same neurons over multiple days or weeks. This can provide insight into how neural activity changes over the course of learning.Anatomical context: Being able to localize imaged neurons in the tissue permits calcium imaging to be combined with other anatomical modalities. Following calcium imaging experiments with electron microscopy or spatial transcriptomics can link physiology with connectivity or transcriptomically defined cell types.
Two-photon calcium imaging#
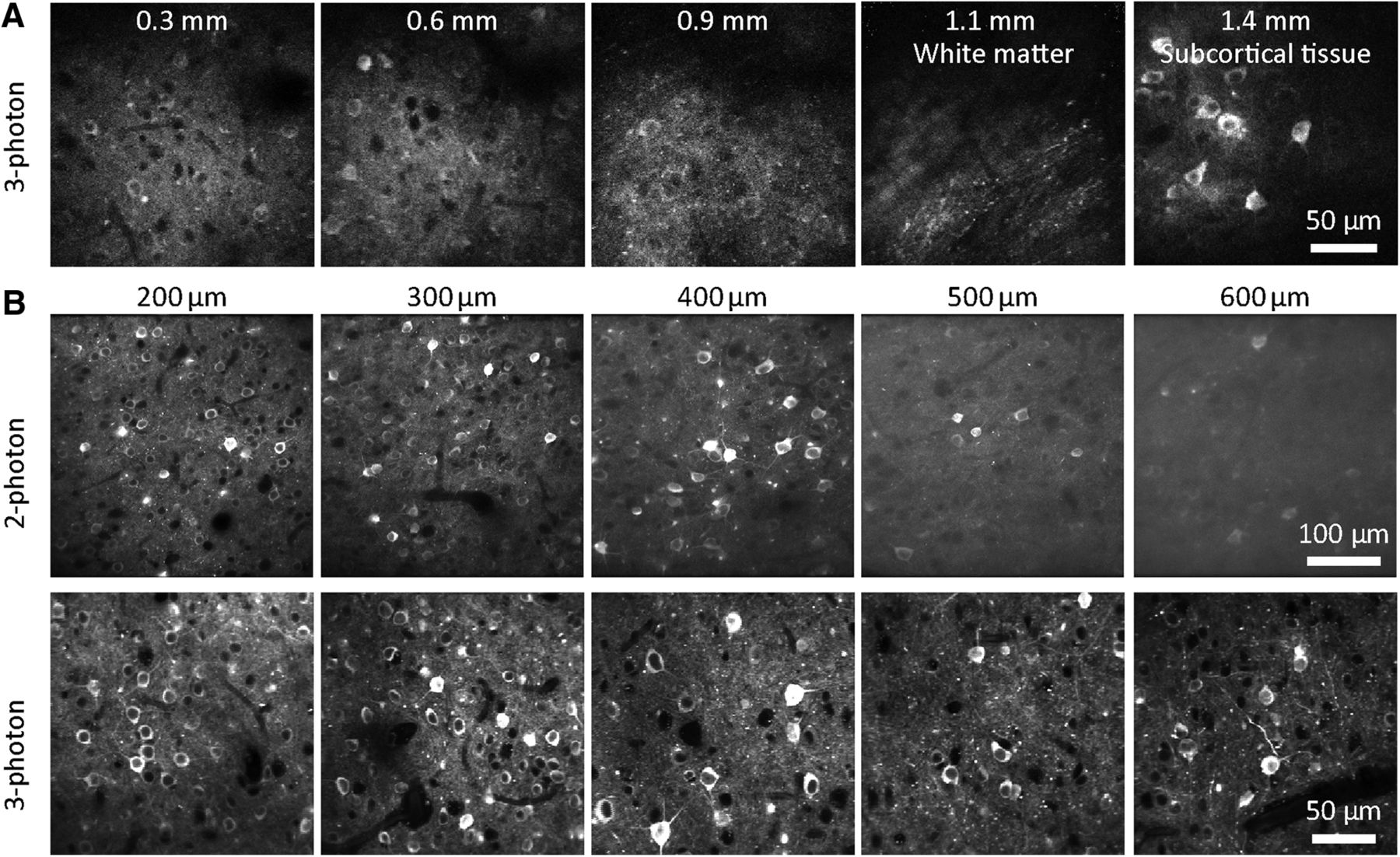
Brains are three dimensional, so microscopy methods that can resolve fluorescence in three dimensions improve our measurements of neurons in intact brains. While wide-field microscopy can still be used on three-dimensional structures, contrast and resolution are hampered by scattering and out-of-focus fluorescence. Confocal imaging rejects out-of-focus light (i.e. provides optical sectioning), but excites large amount of tissue that is not being imaged, and also rejects light that is emitted by the tissue being imaged but is scattered by the intervening tissue. In contrast, two-photon excitation microscopy, or two-photon microscopy for short, uses two photons with longer wavelengths than the emitted light to excite a fluorophore [Denk et al., 1990]. With two-photon excitation, the absorption is a nonlinear function of photon density, allowing microscopes to only excite a single voxel of tissue at a time. This improves our ability to image the activity of GECI expressing neurons in intact tissue.
There are limits to what we can see using two-photon miscroscopy, however. Two-photon excitation generates fluorescence primarily from the focal plane, but out-of-focus fluorescence from outside the focal plane will contaminate the view. This out-of-focus fluorescence increases as the focus is pushed deeper into tissue [Takasaki et al., 2020]. This can be problematic when imaging densely expressed GECIs where there is fluorescence throughout the tissue, where the image quality degrades with imaging depth. However, using Cre lines that limit the GECI expression only to deep neurons (e.g. Cre lines specific to layer 5 or layer 6 pyramidal neurons) can avoid this problem.
Event detection#
The spatial and temporal resolution of the imaging conditions determine the ability to resolve individual, or even small numbers of, spikes. Validation datasets, in which individual neurons are imaged using 2-photon calcium imagine at the same time as intracellular voltage recordings are made using patch clamp techniques, usually collect data with high spatial and temporal resolution. At these resolutions, single action potentials are discernable from the fluorescence traces. However, population recordings like the ones in our ophys datasets, are routinely made with lower spatial and temporal resolutions. This allows us to image large populations of neurons. But, the result is an inability to reliably resolve low firing rate events [Huang et al., 2021].
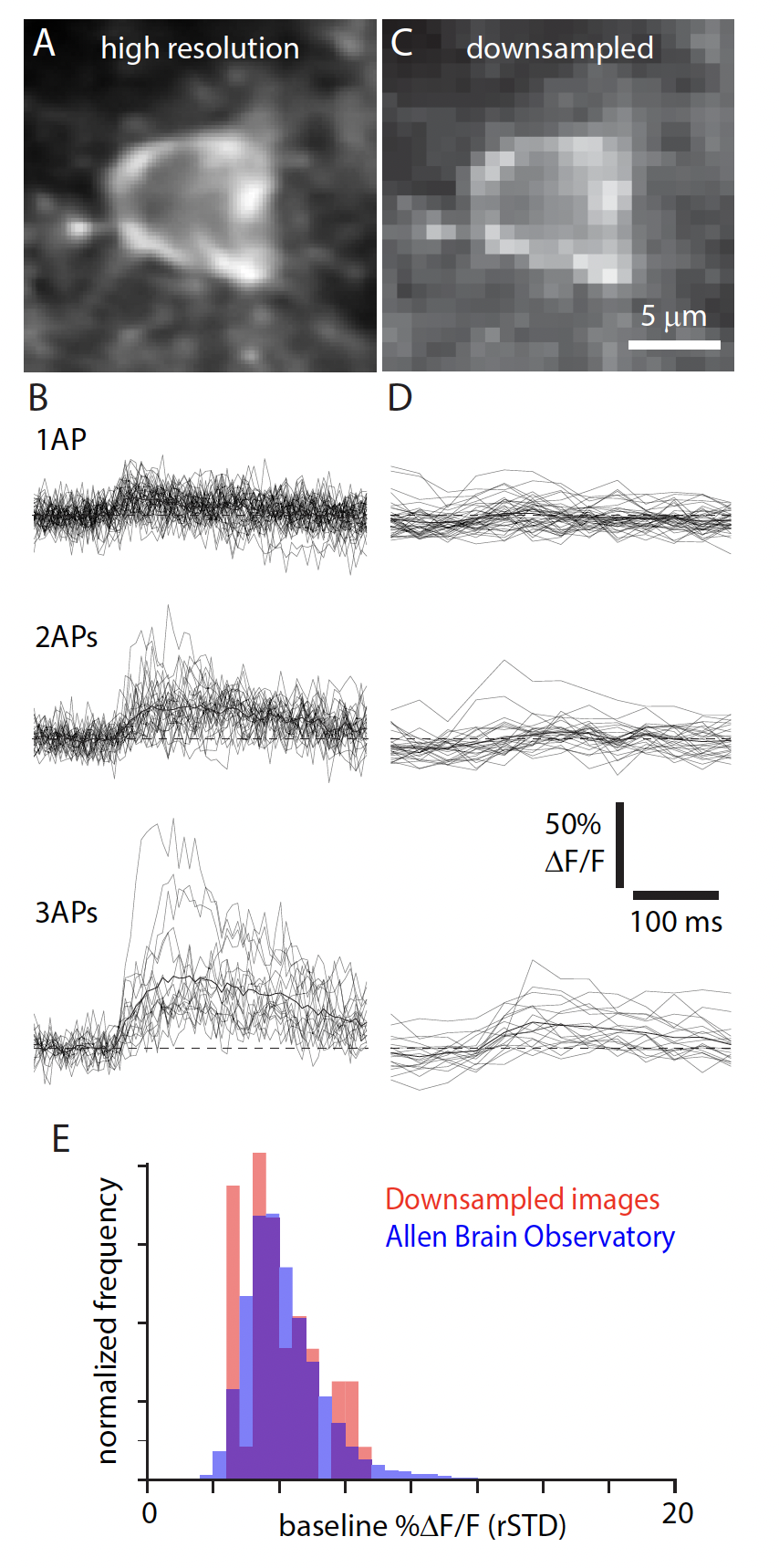
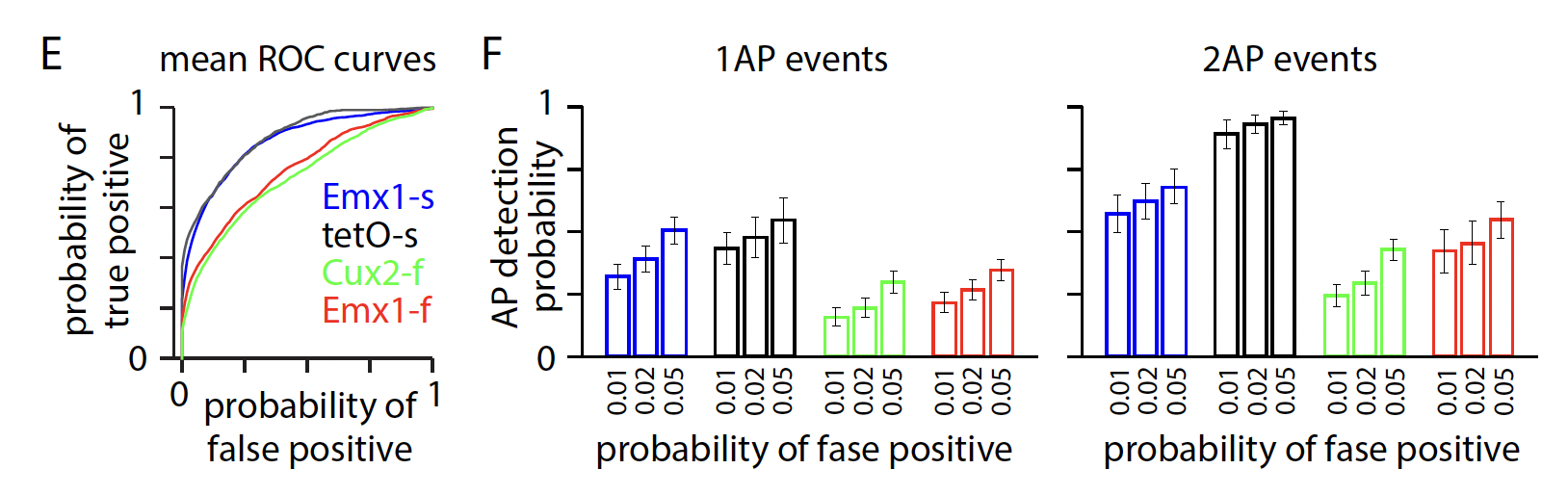
There are several algorithms for detecting “events” based on the DF/F traces. We used a L0 method described in [Jewell and Witten, 2018]. At the downsampled imaging resolution, the ability to detect 1- or 2-AP events is relatively low, particularly for data collected with GCaMP6f.
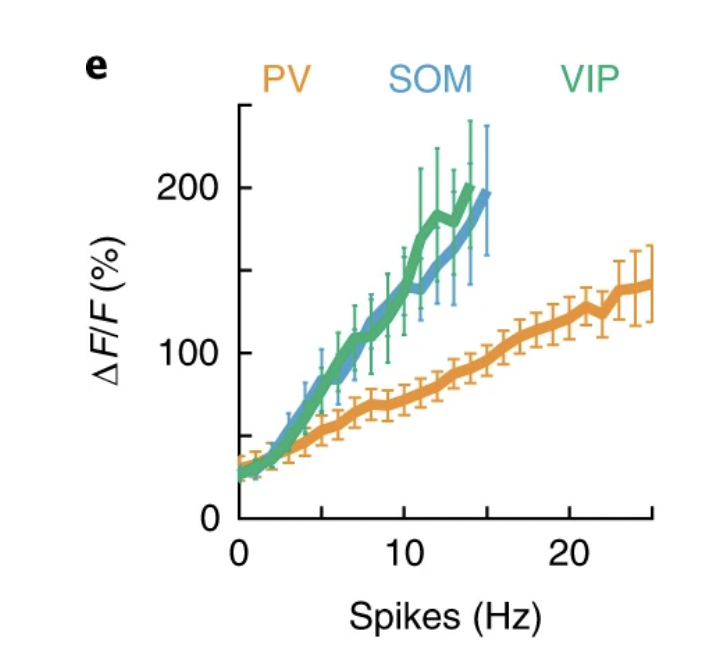
The validation dataset above examines closely the relationship between extracted events and firing rate for excitatory neurons and how that depends on the spatial and temporal resolution [Huang et al., 2021]. This relationship is different for inhibitory Interneurons. Interneurons have a different relationship between fluorescence and firing rate than excitatory neurons. Moreover, this relationship between fluorescence and firing rate is different for parvalbumin-positive interneurons than it is for Somatostatin cells and VIP cells [Khan et al., 2018]. Parvalbumin is a calcium buffer and calcium imaging data of these neurons should be considered judiciously. While general increases/decreases in activity are reflected in the fluorescence signal, analyses focused on more precise event rate estimates and temporal precision should likely be avoided for these PV neurons.